近期,美国 Wessman LL 博士在 JAMA Dermatology 杂志上刊文,报道了 1 例全身坏死性结节、斑块的病例,诊断及治疗存在一定难度,具体如何一起来看看吧。
病例介绍
女性,50 岁,表现为全身散在鳞屑性斑块,部分糜烂、结痂,累及约 10% 体表面积,无浅表淋巴结肿大。组织活检示表皮内有核大深染的不典型淋巴细胞浸润,免疫组化示:CD3+,CD4-,CD8+,真皮乳头纤维化。T 细胞基因重排提示克隆峰。流式细胞术无异常。予以外用皮质激素加窄谱 UVB 光疗,效果欠佳后加用口服贝沙罗汀。
两个月后,皮疹加重,进展为孤立性肿瘤样结节并伴有糜烂、坏死(图 1.A)。行第二次活检(图 1B-D)示亲表皮的淋巴细胞,CD3+,CD4-,CD8+, 颗粒酶 B、TIA-1、B 因子 1 均阳性,CD30、CD56 均阴性。
使用苯丁酸氮芥治疗后有一定缓解,后出现治疗抵抗,皮疹进一步加重(累及约 80% 体表面积),并继发多种感染,故停用苯丁酸氮芥,加强抗感染治疗,并开始使用脂质体阿霉素治疗。
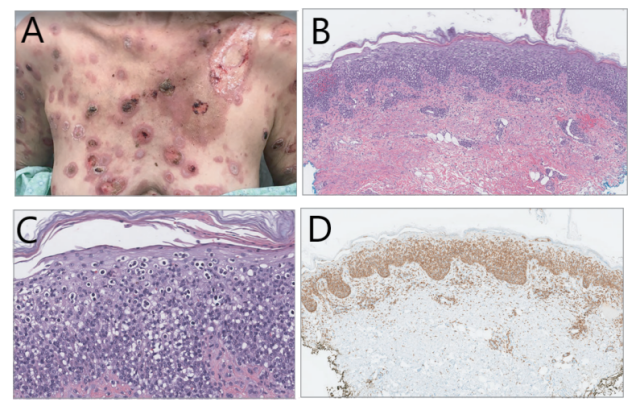
图 1.(A)多个孤立性结节、斑块、糜烂、坏死;(B)示亲表皮的淋巴细胞(HE 染色,50×);(C)核大深染亲表皮的淋巴细胞,未形成典型的 Pautrier 微脓疡(HE 染色,230×);(D)CD8 弥漫阳性(50×)。
综上,您的诊断是什么?
A. CD8+ 蕈样肉芽肿
B. 淋巴瘤样丘疹病,D 型
C. 原发皮肤侵袭性亲表皮细胞毒性 CD8+ T 细胞淋巴瘤
D. NK/T 细胞淋巴瘤
总结
第二次组织病理学分析示核大深染的不典型亲表皮淋巴细胞,其中 CD3、CD8、TIA1、颗粒酶和 B 因子 1 阳性,CD4、CD30、CD56 和 EBER 阴性。支持原发皮肤侵袭性亲表皮细胞毒性 CD8+ T 细胞淋巴瘤(primary cutaneous aggressive epidermotropic cytotoxic CD8+ T-cell lymphoma,PCECTCL)的诊断。
根据 WHO 淋巴瘤分类,PCECTCL 属于原发皮肤 T 细胞淋巴瘤,且占所有皮肤 T 细胞淋巴瘤的比例小于 1%。其特点是突然出现的广泛坏死性斑块或肿瘤样结节,有或无粘膜/内脏受累。
通常认为 PCECTCL 进展很快,而在一些病例报告中,患者最初表现为斑块期 CD8+蕈样肉芽肿(MF),随后缓慢进展为溃疡性肿瘤,数月或数年后才最终确诊。
PCECTCL 的组织病理学改变可以看到明显的网状亲表皮性,通常无 Pautrier 微脓疡形成。浸润的肿瘤细胞表达典型的细胞毒因子:TIA-1、颗粒酶 B、Bf1,不表达 CD2、CD5,极少表达 CD30、CD56。
PCECTCL 与 CD8+MF 的鉴别在于后者病程更长,临床表现更温和。CD8+MF 的显著特征,如色素减退、硬皮病样改变或毛囊粘蛋白沉积,可能有助于区分 CD8+MF 和 PCECTCL,同时 MF 在组织学上更易出现典型的 Pautrier 微脓疡。
淋巴瘤样丘疹病,D 型可表现为丘疹结节,但病程中皮疹时好时坏,且组织学上表现为网状的亲表皮 CD8+T 细胞以及楔形增生的 CD30+不典型淋巴细胞。
NK/T 细胞淋巴瘤则可通过 EBER 来与 PCECTCL 区分。
对于 PCECTCL 需进行全面的实验室及影像学检查以除外潜在的内脏受累。治疗管理包括多药化疗和免疫治疗方案,可考虑造血干细胞移植和全皮肤电子束照射治疗。该病预后差,中位生存期为 12~32 个月。
「文章内容仅用于学术探讨,供医疗专业人士阅读」
