伊藤色素减少症(Hypomelanosis of Ito,HI)是一种神经皮肤疾病,与染色体畸变有关,也被称为无色素性色素失禁症(Incontinentia pigmenti achromians)。近期,韩国首尔国立大学附属医院的 Kim KH 在 Ann Dermatol 上报告了一例多发性先天畸形的 HI 病例,现介绍如下:
病例介绍
患儿女,2 月龄,因不断扩大的色素减退性皮损就诊。患儿在出生时面部、头皮、躯干和四肢即有多发性红色线状糜烂性损害,随后发展为边缘不规则的线状色素减退性斑片(见图 1),无特殊家族史。
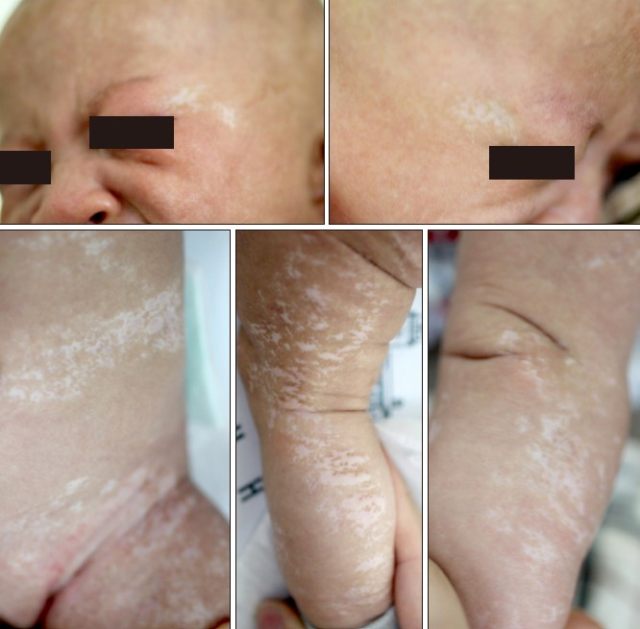
图 1. 面部、头皮、躯干和四肢边缘不规则的线状色素减退性斑片
查体见头皮片状秃发、甲发育不良,眼科检查显示双侧无虹膜、中重度小眼畸形和左眼角膜中央混浊(见图 2A)。肌肉骨骼系统检查显示并趾(见图 2B)、手指发育不良和弯曲畸形以及尾骨偏斜。
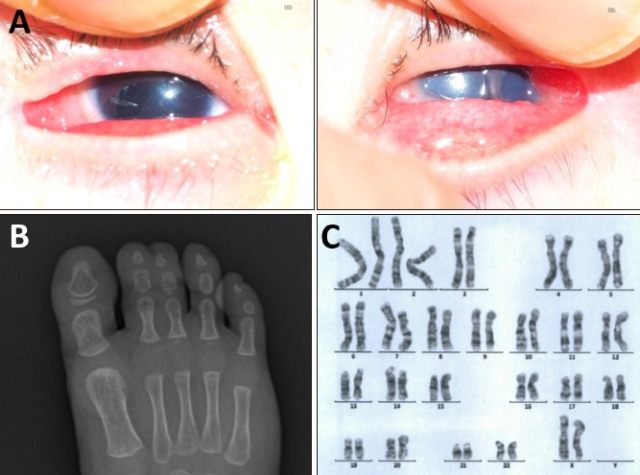
图 2.(A)眼科检查示双侧无虹膜和左眼角膜中央混浊;(B)左第二和第三趾并趾;(C)细胞遗传学分析正常
外周血白细胞染色体分析显示核型正常(46,XX)(见图 2C),IKBKG 基因检测未见突变。结合临床和实验室检查结果,患儿被诊断为 HI 伴多发性先天性畸形。
病例学习
HI 由日本学者在 1952 年首次报告,以沿 Blaschko 线分布的多发性旋涡状、线状或斑片状色素减退-脱失斑为特征。色素性损害在出生时或在出生后数年出现,通常无家族史,多数病例无需特殊治疗。
部分 HI 患者存在系统受累,包括神经系统异常如精神运动发育迟滞、小头或巨头畸形、癫痫发作、张力减退;口腔表现如先天性部分无牙、牙发育不全、圆锥牙;肌肉骨骼畸形如身材矮小、偏侧肥大或萎缩、脊柱侧凸、指趾畸形;眼部异常如斜视、眼球震颤、上睑下垂、角膜混浊等。
HI 被认为是一种皮肤镶嵌现象,本质为「色素性镶嵌」而非一种特殊诊断,遗传学分析显示在包括 HI 在内的多种色素镶嵌性疾病中,色素性基因位点的染色体畸形存在很大程度的重叠,解释了为何不同的镶嵌性表现为相同的表型。HI 沿 Blaschko 线分布的模式可能由胚胎发生过程中表皮细胞迁移和增殖有关。
在临床上,HI 应与色素失禁症和无色素痣相鉴别:
色素失禁症是一种 X 连锁显性遗传病,携带突变 X 染色体的细胞在围产期被清除,产生了色素失禁症的 4 期典型皮损,在出现色素减退前,先经历水疱期、疣状期和色素沉着期 3 个阶段。而 HI 通常为散发性发病,前驱炎症不如色素失禁症显著。
无色素痣大多为孤立型损害,表现为形状不规则的皮肤色素减退;少数为节段型,沿皮节或 Blaschko 线分布;极少数为系统型,累及单侧肢体,呈旋涡状、条索状、泼溅状,与 HI 不易区分。无色素痣通常不伴有神经、肌肉骨骼等系统损害。
「文章内容仅用于学术探讨,供医疗专业人士阅读」
