近期,美国马里兰大学医学院的 Aldana PC 医生在 JAMA Dermatol 上报告了一例以持久性皮肤异色症性斑片伴轻度瘙痒为特征的典型病例,您考虑什么?
病例介绍
患者男,40 余岁,双腋窝和腹股沟皮肤异色症性斑片和薄斑块 5 年,皮损不断扩展至双胁部,自觉轻度瘙痒。患者此前未接受过任何治疗。
查体见色素减退和色素沉着性网状斑片以及萎缩性薄斑块,累及 10% 以上的体表面积,包括双侧腋窝、腹股沟和胁部(见图 1)。颈、腋窝或腹股沟淋巴结无肿大。全面系统回顾结果无殊。
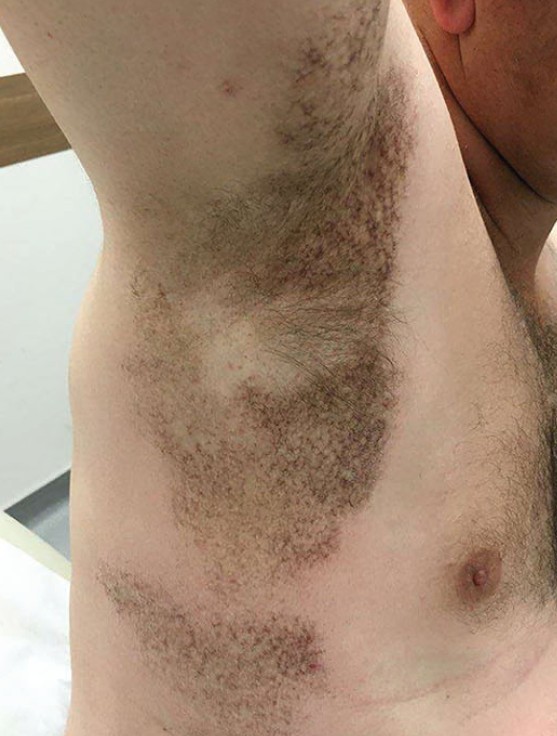
图 1. 右侧腋窝色素沉着性网状斑片伴中央色素减退
血常规、血沉和血生化学结果正常。使用 6 mm 环钻在右胁部进行活检组织学检查,并进行了免疫组化染色(见图 2)。
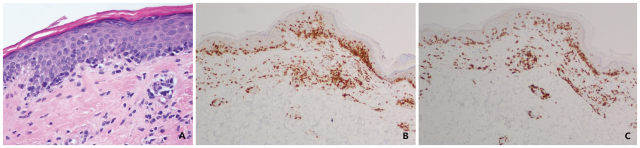
图 2. 右胁部活检组织学检查:(A)HE 染色 200×;(B)免疫组化染色 CD3 阳性;(C)CD8 阳性占优势
结合上述临床和实验室检查,您考虑什么?
A. 蕈样肉芽肿
B. 大斑块型副银屑病
C. 皮肤淀粉样变
D.Dowling-Degos 病
病例学习
组织学检查示浅表血管周围和间质内不典型淋巴细胞浸润,散在累及基底层上方(亲表皮现象),符合斑片期蕈样肉芽肿(MF)。免疫组化示 CD3 弥漫阳性(见图 3B),CD7 表达显著缺失。T 细胞受体(TCR)重排未发现克隆性 TCR 群体。CD8 阳性细胞多于 CD4 阳性细胞(见图 3C)。
结合临床和实验室检查,最终诊断为皮肤异色症性 MF(Poikilodermatous MF,PMF)IB 期病变。患者接受了窄谱中波紫外线(NB-UVB)光疗,皮损显著消退。
PMF 是 MF 的罕见变型,好发于躯干和主要屈侧部位,皮损特征为大片红斑和斑块,含有色素减退和色素沉着区域,伴有红斑、萎缩、轻度脱屑和毛细血管扩张,通常无症状或轻度瘙痒。
与经典型 MF 相似,PMF 好发于男性,预后良好,但发病年龄更轻,自发缓解的比例更大,进展为肿瘤期的患者更少。
PMF 的诊断依赖病理学检查。组织学特征为真皮乳头不典型 T 细胞浸润伴亲表皮现象。另外,还可见真皮浅层血管扩张、嗜黑素细胞、色素失禁和表皮萎缩。与经典型 MF 不同,Pautrier 微脓疡不常见,免疫组化以 CD8 阳性为主。另外,在 21% 的早期 MF 病例中 TCR 重排阴性。
PMF 的治疗方案取决于疾病分期,包括皮肤导向治疗和系统治疗。一线治疗为 NB-UVB 光疗,其他皮肤导向治疗包括外用糖皮质激素、外用贝沙罗汀、细胞毒性药物、PUVA 疗法和放疗。与经典型斑片期 MF 相比,外用糖皮质激素在该亚型中的疗效不佳。皮肤导向治疗失败患者可使用系统治疗如甲氨蝶呤、联合化疗、口服维甲酸类和α干扰素。
在鉴别诊断方面,大斑块型副银屑病常表现为躯干部位轻度瘙痒的萎缩性皮肤异色症性损害,在组织学上,浸润细胞也含有脑回状核淋巴细胞和亲表皮现象,但免疫组化示 CD/CD8 比例正常, 有 CD7 表达,树突状细胞/朗汉斯细胞标记物 CD1a 表达增加。
皮肤异色症样皮肤淀粉样变是原发性局限性皮肤淀粉样变的罕见亚型,好发于四肢,特征为皮肤异色症样皮肤改变、苔藓样丘疹和水疱,组织学可见淀粉样物质沉积。
Dowling-Degos 病呈常染色体显性遗传,通常始于腋窝,逐渐扩展至其他间擦部位,表现为皮肤皱褶部位网状色素沉着和瘙痒。组织学特征表皮突下延,基底层色素增加,乳头上方表皮变薄,无不典型淋巴细胞浸润。
