肢端皮肤剥脱综合征(acral peeling skin syndrome,APSS)是一种少见的常染色体隐性遗传病,由 Fox 医生在 1921 年首次报告,临床特征为肢端皮肤浅表剥脱伴水疱形成,常被误诊为大疱性表皮松解症或剥脱性角质松解症等。
病因与发病机制
多数 APSS 患者存在转谷氨酰胺酶-5(TGM5)基因突变,导致转谷氨酰胺酶-5 的交联活性受到抑制,影响表皮的终末分化和角化细胞膜的形成。
临床表现
APSS 好发于手足背部,表现为无症状性皮肤脱屑,常在受热、浸渍或轻微机械创伤时加重。在儿童患者中常有掌跖水疱形成和糜烂,受累部位不局限于手足背(见图 1~2)。
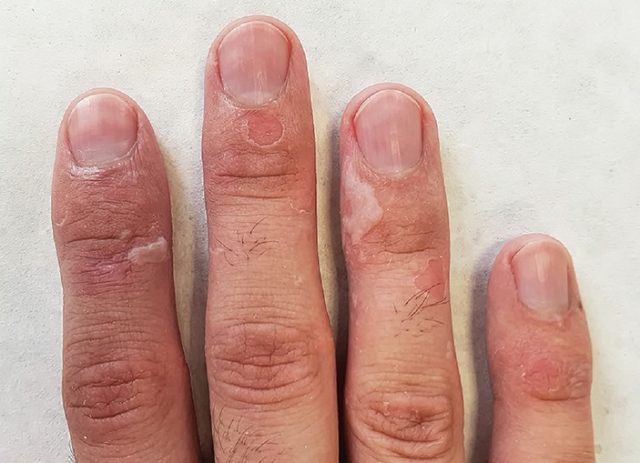
图 1. 指背薄壁水疱和糜烂(图源:Sticova E 等 2019)
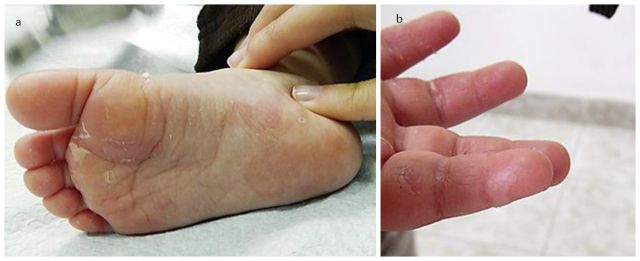
图 2.(a)患儿足底红斑和皮肤剥脱;(b)右示指掌侧松弛性水疱(图源:Kavaklieva S 等 2013)
组织病理
APSS 的组织病理改变为轻度角化过度、表皮在颗粒层与角质层之间或角质层内分离(见图 3)。
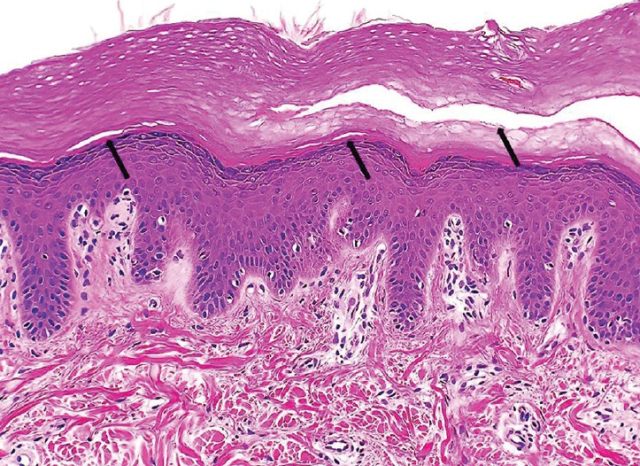
图 3. 颗粒层与角质层之间以及角质层内分离(HE 染色,200×)(图源:Sticova E 等 2019)
诊断与鉴别诊断
APSS 的诊断主要依据个人史以及肢端皮肤浅表剥脱和起疱的临床表现,在临床上,APSS 还应与其他水疱性和剥脱性疾病相鉴别,如局限型单纯型大疱性表皮松解症、角质溶解性冬季红斑、剥脱性角质松解症、手癣、变应性接触性皮炎和汗疱疹等。
APSS 患者的肢端皮肤剥脱症状持续终生,无特应性病史,无前驱变应原或刺激物接触史,斑贴试验和微生物检查结果阴性,特征性组织病理检查有助于确诊。
有学者认为 APSS 的发病率被显著低估,本病常被误诊为局限型单纯型大疱性表皮松解症(见图 4)。在诊断存疑时,分子遗传学检测可作为一项支持性方法,本病由 TGM5 基因突变所致,而单纯型大疱性表皮松解症由 KRT5 和 KRT14 基因突变所致。
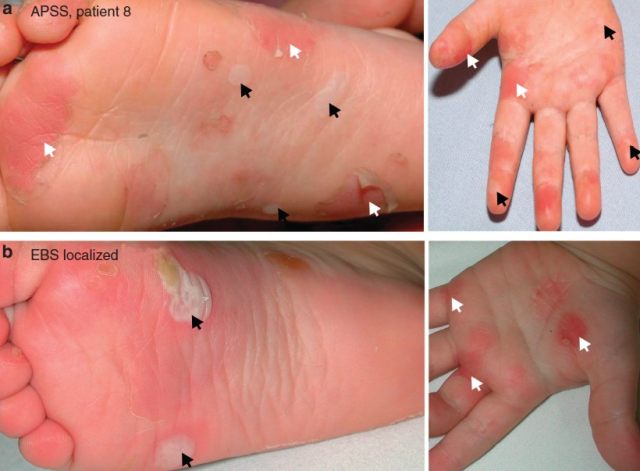
图 4.(a)APSS 与(b)局限型单纯型大疱性表皮松解症患儿可表现为相似的掌跖水疱和糜烂(图源:Kiritsi D 等 2010)
治疗
APSS 的治疗以对症处理为主,包括避免过热、潮湿、受压、摩擦和外伤。外用润肤剂和角质溶解剂对部分患者有益。有人尝试使用甲氨蝶呤、中波紫外线光疗、异维 A 酸和糖皮质激素治疗,但无效。
「文章内容仅用于学术探讨,供医疗专业人士阅读」
参考文献
[1] Sticova E, Kveton M, Dubska M, et al. Acral peeling skin syndrome: An underdiagnosed skin disorder. Indian J Dermatol Venereol Leprol, 2019.
[2] Kavaklieva S, Yordanova I, Bruckner-Tuderman L, et al. Acral peeling skin syndrome resembling epidermolysis bullosa simplex in a 10-month-old boy. Case Rep Dermatol, 2013, 5(2): 210-214.
[3] Kiritsi D, Cosgarea I, Franzke CW, et al. Acral peeling skin syndrome with TGM5 gene mutations may resemble epidermolysis bullosa simplex in young individuals. J Invest Dermatol, 2010, 130(6): 1741-1746.
